Aritalab:Lecture/Biochem/Protein

|
タンパク質は20種類のアミノ酸が連なったポリペプチドです。
タンパク質を大きく分けると表のようになります。
| 水溶性タンパク | 球状 (globular) | 酵素や転写因子など。分子内部は疎水性、外部は親水性。 |
| 不定形 (disordered) | 真核生物に多い不定構造。親水性アミノ酸が多い。とりわけEPQSRKを含む。 | |
| 膜タンパク | 受容体、トランスポーターなど。疎水性アミノ酸が多い。とりわけ膜貫通領域は疎水性。 | |
タンパク質の構造
タンパク質は20種類のアミノ酸から構成され、アミノ酸どうしはアミノ基とカルボキシル基の間にペプチド結合を形成します。 この結合は共鳴によって二重結合性を帯びるので平面構造をとります。 Cα(アミノ酸の中心炭素)の間に炭素と窒素が1個ずつ入りますが、平面が固定されるのでCα-N結合角(φ ファイ)とCα-C結合角は(ψ プサイ)の二結合ぶんで立体配置を決定できます。
一次構造
ペプチド結合によって連結されたアミノ酸の配列を一次構造といいます。コドンの並び順で最初にくるアミノ酸側がアミノ基末端 (N末端)、最後のアミノ酸側がカルボキシル末端 (C末端) となります。
代表的な一次構造のデータベースには以下のものがあります。いずれも特徴的なアミノ酸配列という形式で機能や構造を表現しています。
二次構造
タンパク質のドメインは、二次構造という単位で理解されます。二次構造は水素結合や分子間力で構成されるため、加熱や pH、尿素などの変性剤、界面活性剤などで変化します。
水素結合のエネルギーは、1結合あたり 2 ∼ 10 kcal/mol と見積もられており、共有結合と比較すると 1/10 以下になります。
二次構造の予測にはニューラルネットワークや隠れマルコフモデルが使われます。
予測ツールではInterProScan がよく知られています。これは既存のツール 10 種以上による予測結果をまとめてE-mailで返してくれるシステムで、メタツールと呼ばれます。おおよそ 80 % 程度の精度で二次構造が予測できると考えられています。
- α-へリックス
アミノ酸が平均3.6残基で右巻きにらせんを巻いた構造です。(左巻きもありますが、数は少なくなります。) n 番目のアミノ酸におけるペプチド結合の -C(=O)- 部分が、n + 4 番目のアミノ酸におけるペプチド結合の -N(H)- と水素結合して形成されます。この一般系を 4-α へリックスと呼びます。 このほか 3 アミノ酸、5 アミノ酸で一周する構造もあり、それぞれ 310 へリックス、π へリックスと呼ばれます。
- β-シート
伸長したアミノ酸が並行に並んだストランド構造です。 シートの間で-C(=O)- 部分が -N(H)- と水素結合して形成されます。 ストランドが同じ向きに並んだ場合を並行 β シート、互い違いに並んだ場合を逆並行 β シートといいます。
- ループ領域、ターン領域
α-へリックスやβ-シートは、特定の構造をとるループや、構造が不定のディスオーダー領域で連結されます。細胞外にある多くのタンパク質では、硫黄を含むシステインどうし間にS-S架橋をつくったジスルフィド結合が立体構造を安定化させます[1]。 また、金属イオンと結合して構造を形成するタンパク質(例. ジンクフィンガータンパク質)もあります。
三次構造
いわゆるタンパク質の立体構造を三次構造といい、原子の座標であらわします。三次構造の形成にはアミノ酸側鎖どうしの疎水結合力が大きな役割を果たし、疎水性アミノ酸がタンパク質のコアを形成します。
Protein Data Bank (PDB) は代表的な立体構造のデータベースで、およそ 10 万のタンパク質構造が登録されています。 正確な立体構造はX線結晶解析やNMRで決定するしかありませんが、進化の観点から、構造既知の配列に似ている配列は、立体構造も似ていると仮定できます。
| アミノ酸の保存率 | 検出できる代表的ソフトウェア | 進化的な考察 |
|---|---|---|
| 30% 以上 | 殆どの配列解析ソフト | 同一祖先由来のホモロジーを持つ |
| 25%近辺 | Blast (E-value 10-4) | トワイライトゾーン。配列解析ソフトの限界 |
| 20%以下 | PSI-Blast (E-value 10-4) | 立体構造比較が必須。 |
四次構造
複数のタンパク質サブユニットの空間配置を四次構造といいます。
二面角
アミノ酸の中心に位置する炭素 (Cα) に注目すると、側鎖の他にそれぞれがペプチド結合に関わる -C(=O)- と -N(H)- が接続しています。 ペプチド結合は平面構造をとりますが、それぞれが -C-Cα 軸と Cα-N- 軸を中心に回転できます。 その回転角度 φ, ψ は ±180 ° の間をとることができ、二面角と呼ばれます[2]。
ラマチャンドランプロット
|
アミノ酸配列は、ペプチド結合における二面角のリスト φ1, ψ1, φ2, ψ2, ..., φN, ψN で表すことができます。 この値を平面に散布図としてプロットしたものをラマチャンドラン (Ramachandran) プロットと呼びます。 ラマチャンドラン (Gopalasamudram Narayana Ramachandran) は、インド出身の生物物理学者です。 ペプチド鎖の特徴をみるのに、各アミノ酸のφ角度とψ角度を平面にプロットする方法を考え出しました。 こうするとへリックスとシート構造がきれいに分離され、タンパク質の構造分類に使えます。 |

|
コンタクトマップ
縦(上から下にN→C)横(左から右にN→C)にペプチド鎖を並べ、Cα原子間の距離が10オングストローム以内であれば色を塗ったものをコンタクトマップと呼びます。 対角線は同一の残基がくるので黒くなります。α-へリックスはこの対角線上に重なる形で表示されます。 平行β-シートは対角線から離れた位置に同じ角度で現れ、逆平行β-シートは、対角線と直行する角度で現れます。
タンパク質構造の分類と測定
ドメインによる分類
タンパク質の立体構造を分類するデータベースには、手作業で構造を分けた SCOP (structural classification of proteins) や CATH があります。
大きく分けると以下のようになります。(図はCATH DBより)
mainly α 
|
mainly β 
|
α and β 
|
few structures 
|
測定方法
- ゲル電気泳動
タンパク質は帯電しているので、ポリアクリルアミド電気泳動 (PAGE) で分離できます。
- X線回折
タンパク質を結晶化させられる場合はX線結晶解析により原子位置を特定できます。 粒子加速器から得られるX線を用いると、Cαほか、重い原子の位置を計算できます。水素の位置はわかりません。
- NMR解析
NMRとはNuclear Magnetic Resonanceの略で、原子核の磁気共鳴を用いて原子の位置を特定します。
タンパク質の構造計算
Ab Initio構造予測
どのフォルドを取るかあらかじめ決めずに配列から構造を予測します。
- 分子力学 ... エネルギーが低くなる方向に原子を移動して最適化
- 分子動力学 ... 原子をニュートンの運動方程式に従って動かして最適化
- フラグメントアセンブリ ... 9残基など決められたウィンドウ毎に既知構造ライブラリの中から形状を切り出し、ランダムに入れ替えながら構造を最適化。(ランダムに入れ替えるのでモンテカルロ法の一種)
RMSD
RMSD (root mean square deviation) は二つの点集合
 と
と
 (具体的にはタンパク質を構成するポリペプチドのCα座標) の間に定義される距離の平均値という意味で、次の式であらわされます。
(具体的にはタンパク質を構成するポリペプチドのCα座標) の間に定義される距離の平均値という意味で、次の式であらわされます。

例えば PDB に登録されている二つのタンパク質の構造類似度を測るには、ポリペプチドのCα 座標群を回転・平行移動させてできるだけ重ね合わせたときの RMSD を用います。
分子の力場
タンパク質は巨大なため、構造を量子力学の立場で記述するとあまりに複雑です。 そのため Cα 座標のみを用いた力学系で解釈する分子力学がよく使われます。 代表的な分子力場のモデルには CHARMM (Chemistry at Harvard using Molecular Mechanics) や AMBER (Assisted Model Building with Energy Refinement) があります。
まず分子全体のエネルギーを以下のように表します。アミノ酸 N 個の座標を  と表現します。
と表現します。
![\begin{align}
V(r^N) &= \Big( \sum_{i} \frac{k_i}{2} (l_i - l_{i0})^2
+ \sum_{j} \frac{k_j}{2} (\theta_j - \theta_{j0})^2
+ \sum \frac{V_n}{2} ( 1 + \cos (n\omega - \gamma) ) \Big)\\
&+ \sum_{i=1}^N \sum_{j=i+1}^N \frac{q_iq_j}{4\pi \epsilon_0 r_{ij}}\\
&+ \sum_{i=1}^N \sum_{j=i+1}^N 4 \epsilon_{ij} \Big[ \Big( \frac{\sigma_{ij}}{r_{ij}} \Big)^{12} - \Big( \frac{\sigma_{ij}}{r_{ij}} \Big)^6 \Big]
\end{align}](/mediawiki/images/math/a/8/7/a87a77b2bc529037617d59dab9bee97f.png)
最初の項は共有結合によるエネルギーです。さらに、結合距離、結合角度、結合のねじれに関するエネルギーから構成されます。
- 結合距離 (bond distance)
原子間の結合に対して定められる基準距離  から伸縮して生成するエネルギーはフックの法則に従うと考えます。パラメータは
から伸縮して生成するエネルギーはフックの法則に従うと考えます。パラメータは  です。
です。
- 原子価角度 (valence angle)
Cα が接続する角度がねじれて生成するエネルギーも、フックの法則(二次式)に従うと考えます。同様に  がパラメータです。
がパラメータです。
- ねじれ角
ここにおける  は原子の種類や配置によって決まる定数で、二面角
は原子の種類や配置によって決まる定数で、二面角  がパラメータになります。
ねじれ角のエネルギーは結合距離や原子価角度によるものより小さく、二面角は立体構造の中で柔軟に動きうる部分です。
がパラメータになります。
ねじれ角のエネルギーは結合距離や原子価角度によるものより小さく、二面角は立体構造の中で柔軟に動きうる部分です。
二項目がクーロン力で、全ての Cα 間に定義される長距離相互作用になります。電荷  が距離
が距離  だけ0離れたときのエネルギーを計算しています。
だけ0離れたときのエネルギーを計算しています。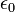 は真空の誘電率です。 最後の項は、ファンデルワールス相互作用で、 Lennard-Jones の12-6ポテンシャルという概念に従うと考えます。
は真空の誘電率です。 最後の項は、ファンデルワールス相互作用で、 Lennard-Jones の12-6ポテンシャルという概念に従うと考えます。
スレッディング
あらかじめ正解の構造がデータベース中にあると仮定して、類似配列を検索する手法です。
- ホモロジーモデリング ... アミノ酸が30%以上保存されるような配列を検索
- 3D-1D法 ... タンパク質の立体構造を考える上で、20種のアミノ酸を区別する必然性は無いことに注目します。各アミノ酸に環境(極性か非極性か、タンパク質の内側か外側か)分類と二次構造のカテゴリーを割り当て、翻訳されたスコアに基づいて配列相同性をみます。
- 解説、参考